
3. THE DIAGNOSIS: GENETIC TESTING
Pamela Magini, Giovanna Cenacchi, Marco Seri
Mendelian genetic diseases are rare clinical conditions caused by a defect in the genome, and are classified as chromosomal disorders, due to either alterations in the number or structure of chromosomes, or gene disorders, when caused by DNA mutations that alter the sequence of genes and, consequently, the structure or function of encoded proteins. Common diseases, whose pathogenesis involves the interaction between genetic susceptibility factors and environmental risk factors, are defined as multifactorial or complex.
Thanks to the completion of the Human Genome Project in the early 2000s and the development of ever more advanced technologies, knowledge on genetic diseases has progressed very rapidly, allowing research in the field of medical genetics to determine the role of specific genes in over 6,000 different diseases (Lander et al. 2001; Venter et al. 2001; International Human Genome Sequencing Consortium 2004; OMIM portal, see some reference links).
The information deriving from the identification of the altered gene, in a specific genetic disorder, has two important clinical applications: therapy and diagnosis. This information can promote the elucidation of the pathogenetic mechanism, responsible for the onset of the disease and the identification of possible targets for specific therapies. To date, however, “curable” genetic diseases are a small percentage, and the most relevant medical application of the knowledge gained from genetic research is diagnosis. In fact, genetic tests are essential for the molecular confirmation of a diagnostic hypothesis based on clinical information, allowing the definition of risk of recurrence, prognosis of the disease, clinical surveillance and possible personalized therapies (ACMG Board of Directors 2015).
Given the importance of the identification of the molecular defect in patients with rare diseases, the use of genetic tests in medical practice has increased exponentially, especially in recent years.
The commonly accepted definition of genetic testing is “the analysis of human DNA, RNA, chromosomes, proteins, and certain metabolites in order to detect heritable disease-related genotypes, mutations, phenotypes or karyotypes for clinical purposes” (Holtzman 1999).
Genetic tests have some peculiarities compared to other laboratory tests. Firstly, their results can have impact not only on the person who has undergone the test, but also on family members. This is why, in common practice, a genetic test must be included within a specific diagnostic path and must be supported by genetic counseling, which gives patients specific indications about its sensitivity and its purposes (Fig. 6).
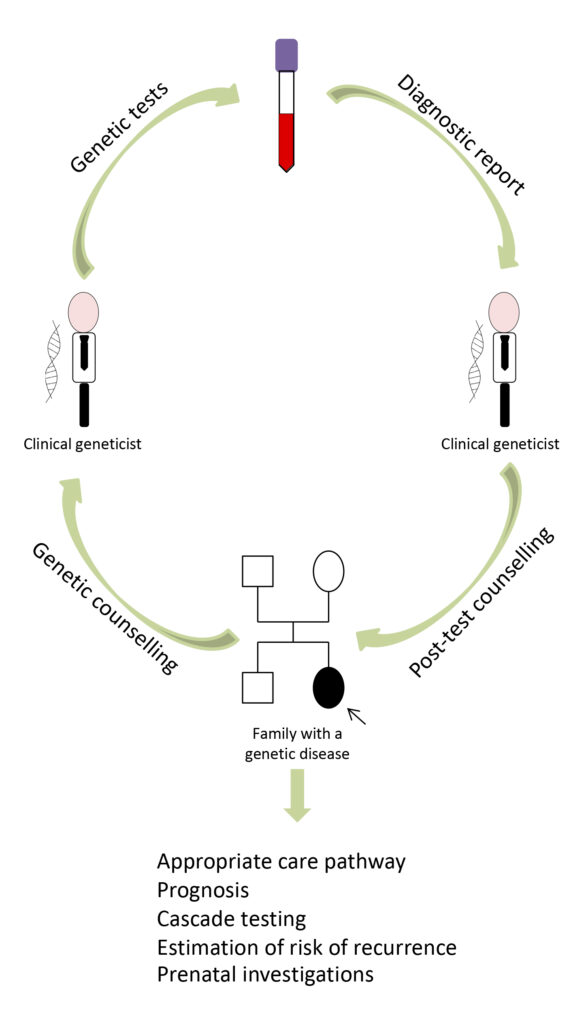
Fig. 6. When a specialist doctor (pediatrician, child neuropsychiatrist, neurologist, etc.) suspects a genetic disorder, he or she generally refers the patient to a clinical geneticist, who provides a physical examination and collects anamnestic data and family history. Based on the data collected during the examination, the clinical geneticist may decide to start a genetic test to confirm a diagnostic hypothesis, explaining the purpose, potential and limitations of the test to the patient or their guardians and collecting informed consent for its execution. After the genetic investigation, the laboratory sends the report to the geneticist who communicates the result to the patient/guardians, during a post-test genetics counselling, together with all the implications for the clinical management of the patient and his/her family.
On the basis of their purposes and the type of disease investigated, genetic tests are classified into diagnostic, presymptomatic, predictive or prenatal (McPherson 2006). Some tests are aimed at evaluating genetic susceptibility for complex diseases, but they often have a limited clinical impact.
3.1. Diagnostic tests
Diagnostic tests allow to establish a diagnosis, or to confirm a suspected pathological condition based on clinical evaluation. With a definite genetic diagnosis, the mode of inheritance of the disease can be determined and thus more appropriate genetic counseling, with specific recurrence risks, can be offered. They can be carried out throughout a person’s life, but also in the prenatal period. Sometimes the outcome of a diagnostic test also allows to evaluate the prognosis of the disease under examination, if the correlation between genotype and phenotype has been characterized thoroughly, so that certain genotypes have been attributed to clinical pictures with well-defined degrees of severity and clinical courses. For example, in most trinucleotide repeat neurological diseases, due to the expansion of specific trinucleotide repetitive elements, the age of onset and the severity of the symptoms are correlated with the number of triplets (Paulson 2018). Furthermore, in some cases, molecular diagnosis can also provide useful information regarding the choice of the most effective therapeutic treatment. For example, the early identification of mutations in the SLC2A1 gene and the timely administration of a ketogenic diet greatly improve the neurological outcome in children suffering from encephalopathy related to GLUT1 deficiency (Wang et al. 2002).
3.2. Presymptomatic tests
Genetic diseases not present at birth but later in life, even in old age, are defined as late-onset. These disorders generally show an autosomal dominant inheritance pattern (due to mutations in one of the two alleles of genes located on chromosomes 1-22, called autosomes) and raise a relevant issue in the diagnostic process. In fact, through the presymptomatic test, the causative mutation previously identified in an affected individual, representing the index case, can be searched for and found in asymptomatic relatives who will inevitably develop the disease throughout their life.
The presymptomatic test is performed without limitations on minors, when the diagnosis can decrease morbidity and/or mortality, thanks to the availability of secondary prevention strategies or adequate therapies. Conversely, the presymptomatic genetic test is limited to adults when results cannot lead to an improved clinical management, as it happens in most cases. People identified as presymptomatic are usually assisted by a multidisciplinary team composed by geneticists, psychologists and appropriate specialists.
However, there are no a priori preclusions for performing the presymptomatic test. The supportive medical assistance will help the patient to understand if results might provide useful information in making choices on certain important aspects of life (maternity/paternity, work, etc.) or, conversely, they could be a great burden to bear, negatively affecting life, when disease symptoms are not yet present. Many diseases, for which presymptomatic tests are available, are neurological. The most classic example is Huntington’s disease (Quaid 2017).
3.3. Predictive tests
The onset of a relatively small group of tumors, called hereditary cancers, such as hereditary breast and ovarian cancer or Lynch syndrome (predisposition to develop non-polyposis colon cancer), is due to mutations of single genes (Garber, Offit 2005). However, in the vast majority of cases, a hereditary alteration in a gene associated with cancer represents only one of the potential factors involved in the development of the disease and is associated with a greater predisposition to the disease. A critical point is therefore the evaluation of the predictive value of the genetic test.
Predictive tests are particularly important, since the possible identification of healthy subjects with a high genetic risk of cancer involves the need to decide whether to take preventive measures. However, the approach to prevention is very complex, as the availability of effective measures varies greatly depending on the type of disease.
3.4. Prenatal tests
During gestation, genetic tests can be used to identify genetic disorders affecting the product of conception. Some tests are carried out without specific indications (in particular chromosome analysis in the case of advanced maternal age, when women have a relatively high risk of pregnancies with chromosomal abnormalities), others focus on the specific genetic disease present in the family (mainly when parents are heterozygous carriers of recessive genetic conditions, or in the case of a mother carrying a X-linked defect).
Some examples are: cytogenetic analysis to detect chromosomal anomalies (for example, the identification of trisomy 21 in Down syndrome), sequence analysis of the CFTR gene in fetuses at risk of cystic fibrosis, the identification of trinucleotide repeat expansions in the FMR1 gene in fetuses conceived by a woman carrying a premutation.
3.5. New technologies applied to medical genetics
Over the years, the continuous development of new technologies for DNA analysis has greatly increased the sensitivity and the rapidity in the identification of both numerical/structural anomalies of the chromosomes, and alterations of gene sequences, as shown in Figure 7.
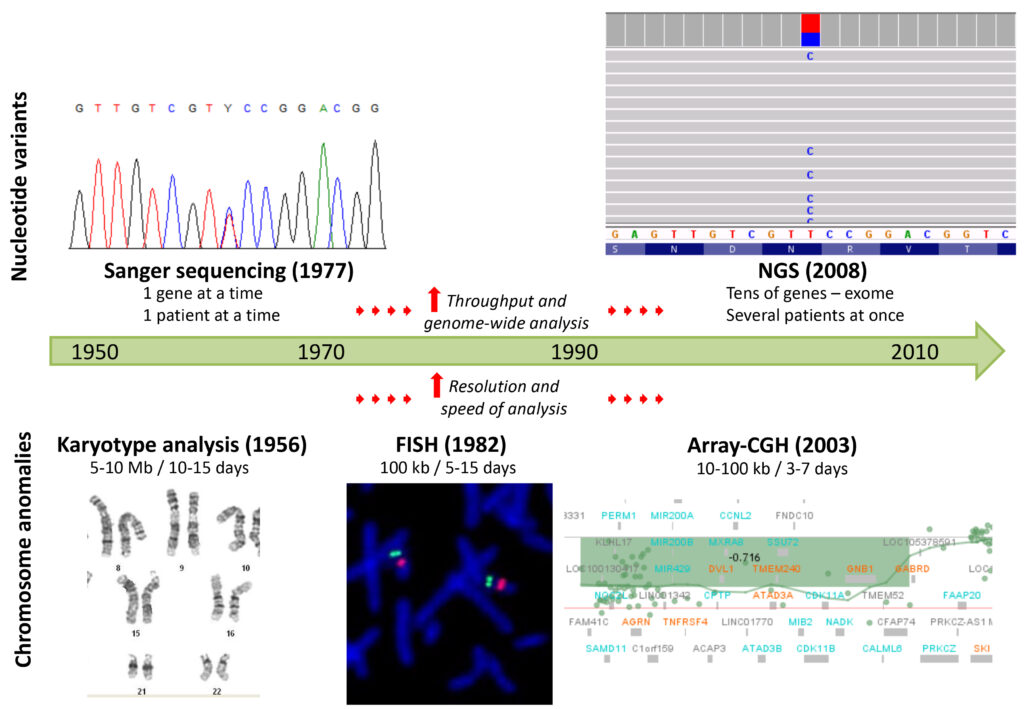
This technological progress has allowed the identification of an increasing number of genes with a role in the pathogenesis of genetic diseases, leading to a significant improvement in the diagnosis within the field of medical genetics (Boycott et al. 2013; Durmaz et al. 2015).
The analysis of the karyotype (classical cytogenetics) allows the detection of large chromosomal anomalies (> 5-10 Mb) through the observation under the light microscope of the entire diploid set of chromosomes (two copies of each chromosome, one paternal and one maternal) of an individual. This was the main diagnostic test from the 1950s to the 1980s, until molecular cytogenetic techniques, such as FISH (Fluorescence In Situ Hybridization), were subsequently developed. By using a fluorescent DNA probe, complementary to the genomic region of interest, FISH is able to detect possible deletions or duplications (Copy Number Variants, CNVs) in the patient’s chromosomes, with higher resolution compared to karyotype analysis (about 100 kb).
The real turning point came at the beginning of the third millennium, thanks to the aCGH (array-based Comparative Genomic Hybridization) technology, in which the patient’s DNA and a reference DNA, labeled with different fluorochromes, compete for the hybridization to oligonucleotides (short nucleotide sequences) complementary to the whole genome (DNA contained in an organism, human in this case), and spotted on a slide. After hybridization, the slide is scanned by lasers, to detect the fluorescence signals, calculate their ratios (patient DNA/reference DNA for each oligonucleotide) and determine the corresponding number of copies, with a high resolution for the identification of CNVs along the whole genome (10-100 kb) and with a few days (3-4) of analysis (Pinkel et al. 1998; Albertson, Pinkel 2003). To date, aCGH is considered the first-tier test for genetic diagnosis in patients suffering mainly from isolated or syndromic intellectual disability, and is frequently applied also in the prenatal period with specific indications, for example when ultrasound anomalies are detected, or for the definition of anomalies identified by karyotype analysis (Park et al. 2011).
Sequencing technologies for the analysis of the nucleotide sequence of genes underwent an even faster development (Heather, Chain 2016). From the second half of the 1970s until a few years ago, the Sanger method was the gold standard sequencing technique, widely used in laboratories. Based on capillary electrophoresis separation of nucleotide fragments, amplified from patient’s DNA and modified through the incorporation of di-deoxyribonucleotides, defined as chain terminators, the Sanger method allows the analysis of the sequence of single disease- genes and the identification of anomalies (nucleotide substitutions, deletions, insertions) with a limited throughput (only one gene at a time). For this reason, the diagnostic process was almost prohibitive, especially for disorders with high genetic heterogeneity, in which several genes are involved. Often, after months of waiting, few genes are sequenced and the genetic cause is not identified.
The development and clinical application of Next Generation Sequencing (NGS) technologies in the first decade of the 2000s, have greatly improved the diagnosis of genetic diseases, both in terms of efficiency and efficacy. These technologies allow the sequencing of multiple genes simultaneously, decreasing significantly the time and costs of the diagnostic process, and offering the patients the concrete possibility of identifying the genetic defect underlying their disease (Jamuar, Tan 2015).
From the analysis of small panels of genes in disorders with well-characterized molecular bases (e.g. chromatinopathies, rasopathies, hereditary spastic paraparesis), the application of NGS has been moved on the sequencing of all known disease-genes (Clinical Exome), which are about 4,000, or even of all the genes included in the human genome (Whole Exome Sequencing, WES) (Xue et al. 2015).
Target differences between the two mentioned platforms, Clinical Exome and WES, give them different potentialities. Since the finding of pathogenic mutations in known disease-genes, analysed by both platforms, has diagnostic value, both the clinical exome and WES can be used as genetic tests. On the other hand, only WES can identify sequence variants with possible clinical significance in genes not yet associated with genetic diseases, thus allowing to improve knowledge in the field of medical genetics, and to outline new genotype-phenotype associations to be included in the clinical diagnostic pathways.
Considering that each individual has tens of thousands of nucleotide variants that have no clinical consequences, analysing all the genes included in the genome in a patient with a genetic disease, and identifying the causative mutation, require a considerable interpretative effort. To facilitate the identification process, various strategies have been implemented, including the simultaneous analysis of parents and the consultation of public databases that collect benign and pathogenic human genetic variants (for example: gnomAD, dbSNP and ClinVar; see some reference links). An additional difficulty concerns the need to demonstrate that the variant, identified in a gene not associated to diseases, is actually responsible for the clinical picture observed in the patient under examination. The effect of the variant on the structure/function of the protein encoded by the altered gene, on cell morphology/homeostasis, or on the development/functionality of organs and organisms can be investigated through in vitro and in vivo functional studies. The sharing of anonymized genetic and clinical data with other laboratories, through dedicated online platforms (for example, GeneMatcher), allows in some cases to identify unrelated individuals with similar phenotypes and variants in the same gene, providing further evidence to support a role of that gene in the pathogenesis of the disease (Quintáns et al. 2014).
3.6. Conclusions
Genetic tests are an essential component of the diagnostic process of people suffering from genetic disorders. Although its therapeutic impact is still limited, the identification of the pathogenic mutation is very important for the clinical management of patients and their families, especially if it is rapid. The use of new technologies, in addition to improving the sensitivity for the identification of CNVs and sequence variants, has considerably shortened the time required for the execution of genetic tests, thus increasing the clinical utility of molecular diagnosis in the context of medical genetics.
In the near future, new sequencing techniques, able to sequence long fragments (long-reads, about 10 kb) suitable for the identification of structural chromosome alterations and CNVs, will probably replace classical cytogenetics and aCGH, and will be combined with NGS (short-read sequencing, up to 150 bp) to offer patients the concrete possibility of arriving at a definitive diagnosis in a short time, undergoing only two tests investigating the entire spectrum of possible genetic and chromosomal alterations.